

| 病名 | 重症肌無力(外科) |
重症肌無力是累及神經肌肉接頭處,突觸後膜上乙酰膽鹼受體的自身免疫性疾病,由於接頭處傳導的安全係數降低而致骨骼肌易疲勞和無力,臨床表現多樣,除橫紋肌無力外尚有內臟表現,使病人喪失勞動力,甚至死亡。
bubble_chart 發病機理
自1960年提出重症肌無力是一種自身免疫性疾病以來,α金環蛇毒素結合的研究發現乙酰膽鹼受體數量減少,伴微小電位波幅降低和病情加重,在90%重症肌無力病人血清中測出獨特的抗乙酰膽鹼受體的抗體,因崦注意至此抗體在重症肌無力發病機制中的作用,設想此抗體減少有功能的乙酰膽鹼受體,從而損害神經肌肉傳導,其機制有:①它加速突觸後膜上乙酰膽鹼受體的退化。②高速或加速受體和抗體複合物細胞內的退化。③減少合成乙酰膽鹼。④它與受體結合後,佔領空間,妨礙受體與乙酰膽鹼結合,加速受體的降解和破壞;通過調理素作用,選擇性地破壞具有受體的公安廳觸後膜。此外,實驗性研究也發現此抗體在重症肌無力發病機制中的作用:①重症肌無力病人血清中有一種降低的體 液因子,用蔗糖遞度沉澱和離子交換色層分析法證明此因子為免疫蛋白IgG、IgG3。②把病血清中的IgG注入實驗動物,可使其微小終板電位波幅降低,神經肌肉接頭處乙酰膽鹼受體減少。③把病人血清注入嚙齒動物,則重症肌無力可被動轉移給此類動物,實驗的過敏性重症肌無力還能從一動物被動轉移給另一動物。上述事例說明抗體在發病機制中對受體的破壞起著重要作用。但也不些研究發現:胸腺切除後的病人,血清中抗體的滴度從未轉為陰性;血漿轉換後,血清中抗體滴度的下降是短暫的,而臨床症狀的緩解可持續數周或數月。因此認為,血清中抗體滴度與臨床症狀并無直接關係,這可能是抗體滴度水平并不反應機體在神經肌肉接頭處的活性。
補體也可能在重症肌無力發病機制中起作用。1977年Engel展示IgG和C3補體沉積在突觸後膜有乙酰膽鹼受體分佈的節段和突觸間隙退化接合褶的碎片上,這可能是抗體與受體結合後激活被 體,破壞突觸後膜,進一步發展到突觸後膜由補體介導的溶解。
最近,Sahashi採用免疫過氧化物酶的方法展示出現C9在突觸後接合褶和突觸間隙,類似C3補體的分佈區。激活C9補體致不可逆的膜破壞,這支持補體介導的溶解在重症肌無力膜破壞機制中的作用,這不同於其他情況,例如Ducenn脊髓病性肌萎縮,它的接合褶被破壞,并沒有IgG或Cq補體參加。最近資料指出,胸腺可能激活補體的替代途徑并加速此反應的過程,抗體也可能使受體引起補全倡導的溶解。體液免疫機制很重要,但在重症肌無力發病機制中仍不能排除細胞介導的免疫機制。實驗性研究展示,在後期發作的病人,其周圍循環血中的T細胞減少,主要減少T細胞的亞類3AI和OKT4。切除胸腺後。這些變化恢復正常。
重症肌無力免疫學的病因尚無定論,自身免疫性疾病多發生在遺傳的基礎上,遺傳柯能為其內因;在外因中,多數人認為與胸腺的慢性病毒感染有關。具有HLA-A1、A8、B8、B12、Dw3、的重症肌無力病人多為女性、青年起病,胸腺增生、無腫瘤、乙酰膽鹼受體抗體檢出率低,用抗膽鹼酯酶藥無效,早期切除胸腺療效好。具有HLA-A2、A3的症肌無力病人多為男性,40歲以後起病,多合併胸腺瘤,乙酰膽鹼受體抗體檢出率高。上述提示遺傳因素有重要作用。
胸腺因素:本世紀初,書籍胸腺與重症肌無力無關,近50年,胸腺切除的資料說明胸腺因素在發病機制中起重要作用。
胸腺的慢性、持續性病毒感染(胸膜炎),使其上皮細胞變成具有新抗原性的肌樣細胞,它們與胚胎肌樣細胞,它們與胚胎肌細胞很相似,是成熟淋巴細胞的前輩,胸腺B淋巴細胞增多,使B淋巴細胞出現異常,導致產生大量自身抗體。這些肌樣細胞具有的乙酰膽鹼受體,其抗原性與橫紋肌細胞的乙酰膽鹼受體有交叉,即由胸腺肌樣細胞新抗原所產生的大量抗,也抗橫紋肌細胞神經肌肉接頭處的乙酰膽鹼受體,於是引起自身免疫性疾病──重症肌無力。已患胸腺炎的胸腺,也可能產生一群T殺傷細胞,破壞神經肌肉接頭處,或生殖出一群T輔助細胞,刺激周圍循環血中的淋巴細胞產生乙酰膽鹼受體抗體。
胸腺切除後症狀緩解的原因,可能除掉了下列來源:①乙酰膽鹼抗原;②乙酰膽鹼受體抗體的產生;③直接攻擊神經肌肉接頭處已致敏的T殺傷細胞;④促使周圍淋巴細胞生產抗體已致敏的T輔助細胞;⑤激活補體途徑導致補體介導溶解的胸腺因素。也不些重症肌無力病例胸腺切除無效,可能由於:①切除不完全;②神經肌肉接頭處的損傷已不可逆;③在胸腺處,位於脾臟和周圍淋巴結中的淋巴細胞群仍引起類胸腺的影響;④長期存活周圍的T細胞仍有活性;⑤異原疾病機制,每位病人對胸腺影響的反應不同。
bubble_chart 臨床表現
重症肌無力在普遍人群中的發病率為1/2萬~1/7.5萬,可發生在任何年齡,但以青年女性和老年男性居多。第一高峰為20歲,第二高峰約50歲,男女比例為1:2,而青年病人中此比例達1:4。橫紋肌無力、疲乏、日重暮輕,活動後加重,休息後減輕為此病的主要症狀。肌無力發作時,每日甚至每小時均有起伏。肌無力可逐漸發作或迅速發作,可完全恢復或部分恢復。首發症狀多為單純眼外肌麻痺,也不單純肢體、延髓肌或頸肌無力者。約56~60%重症肌無力病人眼處肌受累。最後,90%病人均有眼肌無力症狀,表現為上眼瞼下垂、復視,眼瞼下垂在檢查過程中可起伏不定。Cogan症(向上凝視後,提眼瞼上肌下垂)隨著眼肌受累,環眼肌也顯得無力,其他顱神經也受影響,引起吞嚥困難及呼吸困難等潛在的致死併發症。後期發作的病人常損傷咀嚼肌,不能吞嚥,靠鼻飼餵養,舌伸不出口外,肌萎縮且表面有不典型的三條溝。構音困難,聲音低,鼻音重,面肌無力,出現苦笑面容,頸部伸屈肌無力迫使病人以雙手支撐其頭顱。80%以上的病人在眼肌受損1年內發展為全身型肌無力。
四肢肌無力多為對稱性,近端肌群較遠端重,上肢較腿重。個別病人有單條肌肉不對稱的肌無力症狀。深腱反射存在,但重複刺激時可暫時消失。病人常主訴有非特異性感覺,但檢查感覺正常。自主神經系統改變表現為瞳孔改變、膀胱無力和多汗但上述症狀不常見。偶爾發現伴錐體束徵,表現為四肢腱反射亢進,可引起病理反射。精神壓力、狀突然或逐步發作。麻醉或使用肌松劑後重症肌無力表現為頑固的肌無力。
根據病情輕重改良的Osserman分型如下:
Ⅰ型:只有眼肌的症狀和體徵,無死亡率。
ⅡA型:輕度全身肌無力,發作慢,常累及眼肌,逐漸影響骨髓肌及延髓肌。無呼吸困難,對藥物反應差。活動受限,死亡率。
ⅡB:中度全身肌無力,累及延髓肌,呼吸尚好,對藥物反應差。活動受限,死亡率低。
Ⅲ型:急性暴發性發作,早期累及呼吸肌,延髓和骨髓肌受損嚴重,胸腺瘤發現率最高。活動受隱,對藥物治療效差,但死亡率低。
 | 頂新製油 大絕韻 木崗雞蛋 每日C 味全 貝納頌 大醇豆 LCA506 36法郎 Jagabee加卡比薯條 康師傅 順胜實業 頂伸貿易 正義 統一眼鏡 應宏科技 台灣之星 德克士 崑山帆宏 福滿家(廣州市) 全家(中國) |
重症肌無力在各種年齡的臨床症狀各異:
(一)暫時性新生兒重症肌無力
約12~20%患重症肌無力的母親所生的新生兒患重症肌無力,通常出生時即有體徵,但偶爾拖延12~18h,常合併吸吮困難和下嚥困難,哭聲無務,呼吸困難需用輔助呼吸。眼瞼下垂,面肌無務,表情差。母親的乙酰膽鹼受體抗體通過胎盤進入胎兒血中是主要的病因。抗體在胎血中不斷降解破壞,臨床症狀也就相應好轉,故此型稱為暫時性的,其症狀多在3周內自然消失,當逐步減少藥物用量或停藥,未發現復發的危險。對危重的病嬰應立即給予治療,根據病情給口服新斯的明1~5mg,并維持期呼吸功能及營養支持。
(二)先天性重症肌無力
此型指正常母親的新生兒患重症肌無力,家庭中常有重症肌無力病人。42%病例於2歲時,66%於20歲以前發病。病嬰中無乙酰膽鹼受體抗體,其發病機制與遺傳有關。突觸後膜結構畸形;幾乎完全缺乏有功能的接頭褶,微小結構減少,終板乙酰膽鹼受體不足。此型與暫時性新生兒重症肌無力不同,症狀為持續性,無完全緩解。症狀多在出生時或其後不久出現,眼外肌受累明顯,常可累及面部肌肉而影響攝食。全身肌無力少見。
(三)家族性嬰兒型重症肌無力
指正常母親的嬰患重症肌無力,家族中有其他重症肌無力病人,如兄弟或姊妹,為常染色體隱性遺傳出生時有嚴重的呼吸困難和攝食困難,尤以呼吸暫停的特點而有別於前兩型,常因呼吸衰竭致使嬰兒死亡。多在2歲內症狀發作,有自然緩解傾向,隨年齡增長百好轉,但也可因感染後再將近引起窒息致死。抗膽鹼酯酶藥物治療有效,故應早期確診。
(四)膽鹼酯酶缺乏
此型重症肌無力是由於在終板亞神經結構缺乏乙酰膽鹼酯酶所致,發生於兒童累及眼肌和顱神經Ⅸ~Ⅻ支配的肌肉,軀幹肌肉也受累,肢體近端較遠端重。騰喜龍試驗陰性,用抗腫鹼酯酶藥物或增加乙酰膽鹼釋放的胍無效,而強的松治療效果明顯。
(五)青少年肌無力
全部肌無力病例的4%在10歲前發病,24%病例在20歲前發作,女性佔優勢(4:1)。此型與嬰兒型相反,遺傳因素相對小,主要是免疫機制在發病機制中起作用。病程進展慢,有明顯起伏。胸腺瘤少見。
(六)成人肌無力
705成人肌無力病例有胸腺增生,年輕人多見;10~15%病例有胸腺瘤,老年人常見。男性病人較女性發病快,緩解率低,死亡率高。臨床過程有明顯的加劇期和緩解期,3/4眼肌受累的病人,在第1~3年內發展成全身型肌無力,咽喉肌受損,最嚴重時,可有多組肌群受累而出現不對稱的症狀組合。活下來的大部分病人變為慢性遷延,發作次數減少,症狀減輕。
(一)藥物檢查
抗膽鹼酯酶藥物阻滯乙酰膽鹼在突觸裂水解,延長它的作用和增強乙酰膽鹼受體的相互作用的能力,升高微小終板電位,增加神經肌肉傳導的安全係數。這些藥物能緩解或減輕重症肌無力病人的臨床症狀和電生理異常,最常用的抗膽鹼酯酶藥是騰喜龍,它的作用短暫,對95%重症肌無力病例有效。陽性反應則可確診,個別病例反應陰性,但不能排除重症肌無力的診斷。建議傍晚或運動後在肌無力最重時才作此檢查。眼肌對此藥物最不敏感,故對局限於眼肌的重症肌無力病例難以做出診斷。
靜注2~10mg騰喜龍,初量2mg作敏感試驗,為對正服抗膽鹼酯酶藥的病人,避免出現增加膽鹼能的肌無力症狀,作此檢查時,應準備好處理過敏反應和呼吸併發症。陽性反應一般採用三聯盲目的方法,用生理鹽水和煙鹼酸作對照。騰喜龍引起輕度頭痛、發熱感、煙鹼酸可重複某些症狀,但不影響神經肌肉傳導,所以是比較理想的對照藥。
如反應短暫,用常規床旁技術不易作記錄時,可用長效的抗膽鹼酯酶藥物,它們有較長的潛伏期和作用期。新斯的明1.5mg肌注,10~30min內可改善症狀,持續4h。如反應仍不肯定,則可作長期試驗,口報抗膽鹼酯酶藥數周。
(二)電生理檢查
重症肌無力病人的電生理表現為微小終板電位的振幅降低。Jolly試驗是重複刺激一根神經,正常人可以忍受40~50次/s的刺激。在重症肌無力病人,2~3次/s的刺激就會引起活動電位不正常的遞減。國內採用低頻重複電刺激(2、3、5、10及20次/s),發現此方法有診斷意義。上述試驗優點在於簡便,但不十分敏感,特別在發病早期,約50%重症肌無力病人對此檢查并無改變。
檢查神經肌肉傳導較為敏感的方法是單根纖維肌電圖檢查。用單根纖維針電極,插在同一運動神經支配的二根肌纖維之間。兩個活動電位之間潛伏期的各種形式以顫抖來表示。重症肌無力病人神經肌肉傳導的各種形式是顫抖增加或在嚴重病例,阻滯一個活動電位,兩個活動電位之間的潛伏期很長。95%有多組肌肉被累及的重症肌無力病人在檢查時,顫抖不正常,顫抖代表微小終板電位振幅的功能。這個檢查可用作監測重症肌無力的臨床過程。它的優點是較為敏感,能及早作出診斷,但需要使用昂貴的設備和神經生理的評定。
鐙骨反射衰減也被用來診斷重症肌無力對眼型高度敏感,但對全身型反應差。
(三)血清學檢查
重症肌無力病人血清中含有許多非特異性抗體,包括抗橫紋肌、抗核、抗甲狀腺、抗胃壁、抗產生精子和抗神經元的抗體,可測定其含量供診斷作參考。
從眼鏡蛇分離提純出的特殊神經毒──α-金環蛇毒,被 發現有針對性地不可逆轉的凝固乙酰膽鹼受體的活動部位,這個毒素或以識別乙酰膽鹼受體抗并可測出其數量。這個檢查是將被檢驗的血清作用於從人肌肉取得的乙酰膽鹼受體抗原,後者已埋有125I標記的α-金環蛇毒形成一個複合物,它凝固在受體上相鄰的部位。繼之抗人蛋白,沉澱這個複合物。根據沉澱劑上的放射性可計算出乙酰膽鹼受體抗體的水平(放射免疫試驗)。血清中的乙酰膽鹼受體抗體對重症肌無力呈高度特異性,可在90%的病例測出。它只有在服用青霉胺或以蛇毒接種時被發現,而且從損壞的肌肉所釋放出的乙酰膽鹼受體不產生抗體。一般認為此抗體水平與病人的臨床症狀無關,但單純眼型病人傾向於有較低的抗體滴度。
最近採用酶聯免疫吸附試驗(ELISA)測定抗體,其敏感度較放射免疫試驗高。用α-銀環蛇毒素2.5pmol/孔包被。中層為由電鰻電器官提取的乙酰膽鹼受體0.2pmol/孔。同時測定乙酰膽鹼受體的相對滴度;即病人於某一時相的絕對滴度與其最高絕對滴度百分比。經直線回歸和相關性研究,證明重症肌無力病人肌無務程度與其血清中乙酰膽鹼受體抗體相對滴度密切相關。此外,還可進行血清免疫球蛋白檢查,包括IgG、IgA、IgM、C3、補體測定等免疫學檢查,均有助於重症肌無力的診斷。
(四)胸部X線檢查
常規胸部平片仍是目前比較簡單的檢查方法,對胸腺瘤的診斷可達62%。胸腺縱隔區體層攝影可發現30%胸片陰性的病例有胸腺瘤。胸部CT診斷符合率達94%,CT掃瞄可鑒別囊性或實性,有否鈣化並能發現較小的胸腺瘤,也可看出有否侵犯胸膜、肺及大血管等惡性胸腺瘤的指徵。
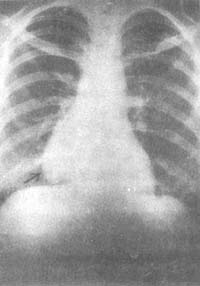
圖1 後前位胸片示:右心緣旁陰影
男,58歲,重症肌無力合併胸腺瘤患者

圖2 右側位胸部體層像示:
前縱隔陰影(同上例)
重症肌無力可合併其他疾病,例如類風濕性關節炎、全身紅斑狼瘡、多肌炎、 Sjogren綜合徵,潰瘍性結腸炎等自身免疫性疾病;合併維生素B12缺乏,甲狀腺疾病,糖尿病,甲狀旁腺疾病,腎上腺疾病和白斑等,被認為是多腺體衰竭綜合徵的一部分。這些可能有遺傳的因素,基於他們與組織相容性的抗原有聯繫,特別是HLA-AI、A8、Dw3,這些成為自身免疫性疾病的危險因素,在某一病人,一次特殊的接觸可引起不正常的反應。這個推論是根據研究單卵雙生同胞胎的資料,發現只有其中一位嬰兒患重症肌無力。
5%重症肌無力病人有甲狀腺功能障礙。有時,難以區分重症肌無力症狀還是甲狀腺疾病的症狀,因為兩者可引起近端腩體無力和眼症。甲狀腺病主要是內分泌疾患,而重症肌無力更多的是免疫性或遺傳性疾病。所有甲狀腺病,包括甲狀腺腫、粘液水腫、Graves 病和橋木甲狀腺炎都可合併肌無力。
1.肌無力綜合徵與重症肌無力的鑒別診斷
肌無力綜合徵(Lambert-Eaton綜合徵)是後天性運動神經末稍的疾病,由於釋放乙酰膽鹼數量減少所致。典型的病人是50~70歲男性,主訴肢體帶狀肌群無力,主要是上肢,而下肢、眼肌或延髓受累較輕或未被累及,深部腱反射傾向於減弱或正常。此病可誤診為重症肌無力,它常伴有腫瘤,特別是小細胞肺癌,肌無力先於腫瘤症狀出現。
肌無力綜合徵有自身免疫的基礎,致病的IgG抗體與突觸前,主要負責釋放乙酰膽鹼的鈣離子系統有交叉反應。在有病的神經終板,乙酰膽鹼含量和持乙酰酶活動度正常,說明乙酰膽鹼的合成和集中是正常的,而缺陷是由於小囊泡釋放受損,減少乙酰膽鹼的釋放量造成此疾病。由於在膽鹼能自行調節部位的乙酰膽鹼釋放量減少,繼發出現家族性自主神經功能異常,表現為嘴乾,損傷眼肌,使眼球對不同距離的調節能力受損,排尿困難和便秘。
肌無力綜合徵的典型肌電圖呈遞減現象。與重症肌無務相反,增加運動量和痙攣性刺激比減小肌力更會減輕此現象。病人通常有面部肌無力的「苦笑鬼臉」,但肌力相對好,而重症肌無力病人面部變化不太重,但肌無力明顯。肌無力綜合徵病人的骨骼肌無力用抗乙酰膽鹼酯酶治療時效差,而3,4-二氨基吡啶證明可增加傳遞質的釋放,對抗神經肌肉和自身免疫的神經系疾病有效。增加從突觸前神經末稍釋放乙酰膽鹼的藥物,如鈣可能有效。
重症肌無力病人結非去極化肌松劑敏感,而對去極化肌松劑耐藥。肌無力綜合徵病人對上述兩種肌松劑均敏感。
在診斷此綜合徵時,應做支氣管鏡檢查及縱隔鏡檢查,如懷疑肺癌應開胸檢查。
2.其他疾病與重症肌無力的鑒別 癔病、甲狀腺疾病、神經肌肉疾病的其他肌無力的症狀有時被誤診為重症肌無力,但氯化騰喜龍試驗、單根纖維肌電圖、抗體水平的測定均可鑒別這些疾病。


